Skip to content
AMSPW
L'annuaire des sites francophones
Navigation
blogueur.net
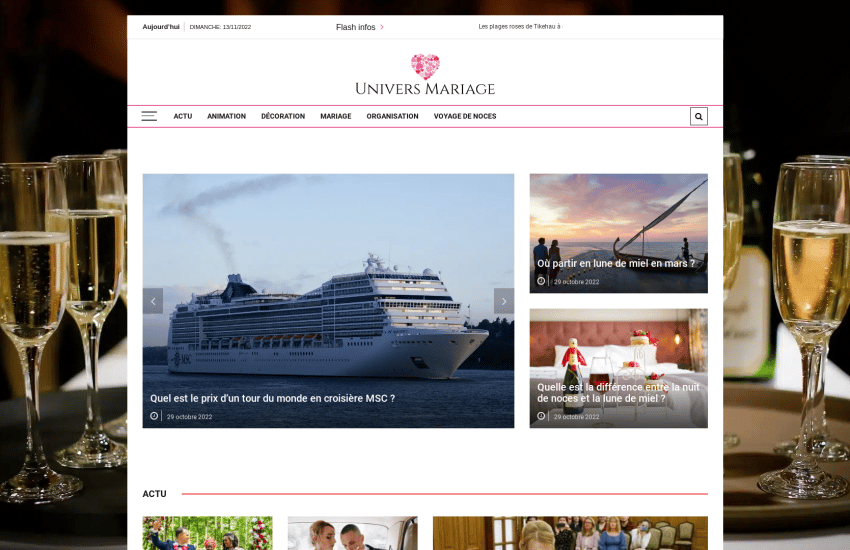
univers-mariage.net
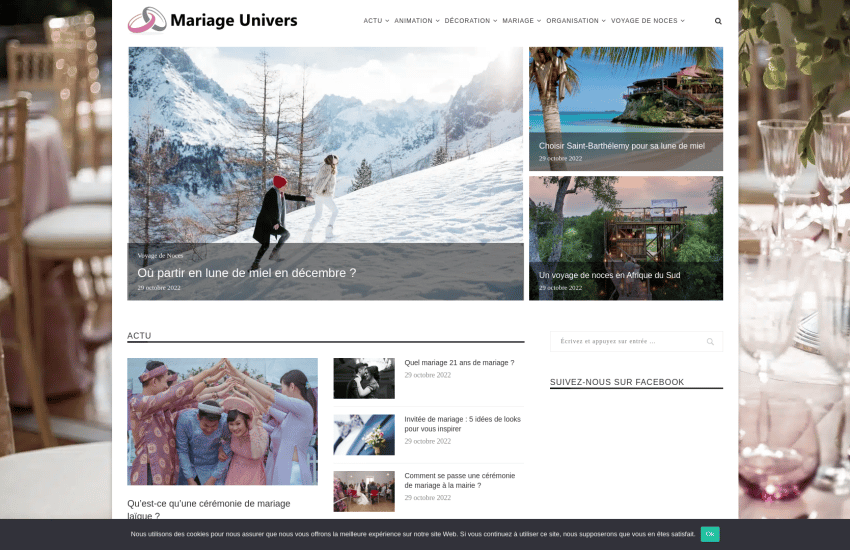
mariage-univers.fr
mariage-conseils.fr
belle-et-unique.fr
actu-buzz.net
lebloginfo.fr
animal-liberation.net
lacavernedugeek.com
must-car.fr
cyberimmobilier.fr
sport-cars.fr
h-immobilier.fr
mariage-magazine.fr
leschiensnefontpasdeschats.fr
planifiez-votre-mariage.fr
la-maison.info
automaniacs.fr
expert-jardin.fr
mamzelleh.com
jobemploi.net
voyage-sur-mesure.com
france-sports.fr
francemedicale.net
225business.com
joliefamily.fr
club-voyageur.fr
msmedical.net
the-click.net
optisante.fr
jardinjade.com
spy-immo.fr
parentsensemble.com
mon-habitat-web.com
hoteantictravel.fr
justindeco.fr
mamanpascommelesautresoupresque.fr
madame-dentelle.fr
car-system.fr
sos-beaute.com
jardinier.net
monde-immobilier.com
s-finance.fr
lebioducoin.fr
tout-immo.net
h2osport.fr
santeducation.com
yoopitravel.com
immersivelab.fr
businessinfo.fr
hucky.org
bretagne-net.com
ecseri.net
consultantweb.net
littlebreizh.fr
u-games.ch
maisonfutureco.fr
jeune-bretagne.com
pole-immo.fr
impact-patrimoine.fr
Navigation des articles
1
2
Next Posts
»